Cystic fibrosis, lungs
English: mucoviscidosis, cystic fibrosis
Cystic fibrosis is an inherited disease. The inheritance is medically referred to as autosomal recessive. Cystic fibrosis (cystic fibrosis) is not inherited on the sex chromosomes X and Y, but on the autosomal chromosome 7.
Read our general article on metabolic disorders: Metabolic disorders - what does it mean?
The mutation is on the so-called CFTR gene. Recessive meant that two defective copies of the gene had to be present for the disease to break out. If a person has a healthy and mutated gene location on the corresponding chromosome 7, the disease does not occur.
$config[ads_text1] not found
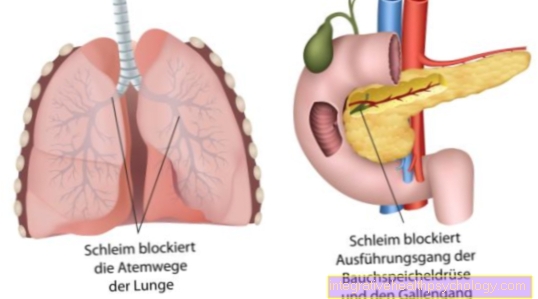
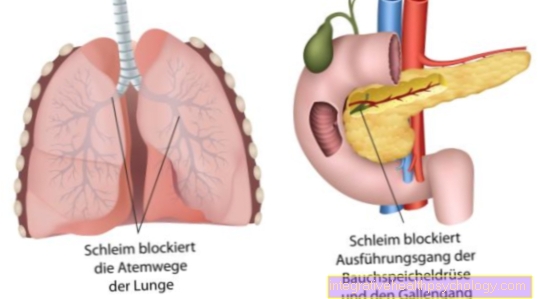
The result is a pathological gene product. The thereby coded Chloride channels are broken. The defective chloride channels lead to the formation of thick mucus in all exocrine glands.
These exocrine glands, i.e. glands that release their secretion to the outside, include:
Cystic fibrosis is a Hereditary disease. It is inherited in such a way that it is gender-independent and only with two defective genes occurs. It is the most common autosomal recessive inheritance.
The consequences are tough mucus formations of all exocrine glands, such as the glands of the lungs, the pancreas and also the sweat glands. They are based on that disturbed transport of chloride between the inside and outside of the cell (read on: Chloride in the blood). The mutated gene is on Chromosome 7 and causes a wide variety of organ involvement with the corresponding effects on breathing, digestion and reproduction.
Unfortunately, therapy can only alleviate the symptoms, but not bring about a cure. The Life expectancy in cystic fibrosis patients relatively low.
Since it is a recessive inherited disease, there are people who carry the changed gene but do not suffer from the disease itself. Such persons are called Feature carrier or Conductors, i.e. carriers. These people do not have cystic fibrosis because the other copy of the gene is intact and the sick one is not strong enough to prevail.
However, she can pass this defective copy of the gene on to her offspring. If a modified gene were already sufficient to cause a disease, it would be a so-called dominant inheritance. Such an inheritance can be found, for example, in Chorea huntington. You can find out more about this disease under our topic Chorea huntington.
At about 1:2500 lies the Disease rate in newborns in Germany. Carrier is about everyone 25. in the German population.

Cystic fibrosis is caused by a mutation of a gene on chromosome 7. This chromosome is an autosomal chromosome, not a sex chromosome.
Everyone has 44 autosomal chromosomes (two identical versions of each) and two sex chromosomes. This mutation on chromosome 7 leads to the formation of defective chloride channels. The reabsorption (re-absorption) of chloride from the glandular secretions is not possible because the receptor, the docking point for chloride, is not built into the gland ducts.
Instead, it is put down for mining due to its incorrect appearance and structure. The natural exchange of chloride through certain chloride channels is disturbed. These so-called channels are made up of proteins. A wide variety of proteins are encoded on our DNA. Due to the genetic defect of the chloride channels, there is a dehydrated and tough production of mucus from all glands, which release their secretion to the outside. The mucus then partially blocks the ducts or the airways in the lungs.
$config[ads_text2] not found
$config[ads_text3] not foundRead about this too Chromosome mutation

The typical symptoms that begin in infancy are groundbreaking in the diagnosis of cystic fibrosis.
This suspicion is reinforced by a positive family history (illness of the father / mother or close relatives). A positive family history means that there are or have already been cases of cystic fibrosis within the family - on the maternal or paternal side.
The lack of pancreatic enzymes can also be detected in the stool. Any blockages in the airways can be detected by x-raying the chest.
A sweat test, which measures the chloride content of sweat, also helps with the diagnosis of cystic fibrosis. If a certain value is exceeded and the other symptoms also apply, the diagnosis is relatively fixed. Often the parents themselves notice the increased salt content in the sweat of the infant.
The unborn child can also be tested for this hereditary disease. Using an amniotic fluid puncture (Amniocentesis) fetal cells are removed and examined for the mutated gene.
Read more on the topic: X-ray examination of the child
Anyone affected by cystic fibrosis will receive advice in one Cystic fibrosis - outpatient department or advice from Human geneticist (Specialist in hereditary diseases) recommended. These can help to increase the quality of life or, if you want to have children, calculate the probability of a sick child. Provided the parents are fertile and fertile.
Otherwise, treatment is symptomatic, as the cause, the defective gene, cannot be eliminated.
$config[ads_text4] not found
Cystic fibrosis (cystic fibrosis) is still an incurable disease today.
In the case of cystic fibrosis, it is important to have an adequate intake of table salt (Sodium chloride, NaCl). Mucolysis is of course aimed at. Mucolysis is the dissolving of mucus, especially in the lungs, to make breathing easier.
Medicines and inhalation can alleviate the symptoms. If the lung function is noticeably deteriorating, oxygen can be given.
Through intensive physiotherapy (physiotherapy), for example tapping massage and breathing exercises, the lung changes caused by cystic fibrosis are also treated.
Often times, the disease ends with a required lung transplant. However, the waiting lists are long.
Oral administration of pancreatic enzymes and fat-soluble vitamins is also part of the therapy. The task of the pancreas must therefore be supported, or rather replaced. Fat-soluble vitamins are A, D, E and K. They must be given directly into the blood as they cannot be absorbed from food due to a lack of digestive enzymes.
The diet should also be high in calories, as only a fraction of them can be obtained from food.
To avoid additional risk factors for complications such as the flu or pneumonia, the child should be vaccinated. The following vaccinations are recommended:
Read more on the topic: Superinfection
Of course, these measures require a consultation with a doctor, with whom the risks should be discussed.
Nowadays, great hope for cystic fibrosis therapy is placed in genetic research. An attempt is made to introduce the missing genetic information into the human genome. We are looking for vectors that can master this task. Vectors can, for example, be bacterial or viral DNA that manage to incorporate the healthy frequency into our genome.
The therapeutic approach in unborn patients is currently being tested. In mice, the mouse embryos have already succeeded in introducing the healthy gene, which contained the correct gene sequence, through amniocentesis (amniotic fluid inoculation). The healthy CFTR gene was thus produced in these mice. Amniocentesis is a puncture and removal of child's cells from the amniotic fluid. This is done through the mother's abdominal wall.
$config[ads_text1] not foundIn Germany, however, this form of intrauterine (= in the uterus = in the womb) "therapy" is prohibited.
A preventive measure in this sense it does not exist because it is a hereditary disease.
However, a human genetic counseling center (usually found in university hospitals) can be visited. Here it is calculated how high the risk would be to pass the disease on to children.
This advice is always useful if there is a family history of cystic fibrosis.
Also one Prenatal diagnostics is worth striving for. Here before the birth (i.e. prenatally) a Amniotic fluid examination (Amniocentesis) carried out. Fetal cells (cells from the child) are taken from the amniotic fluid and the DNA is examined for the mutated gene.
Unfortunately, the mean life expectancy in patients with cystic fibrosis is only 32-37 years. Today, the life expectancy of newborns born with this condition is estimated to be around 45-50 years.
The prognosis depends heavily on the therapy and whether it is adhered to.
The patient himself and his motivation therefore play an important role.